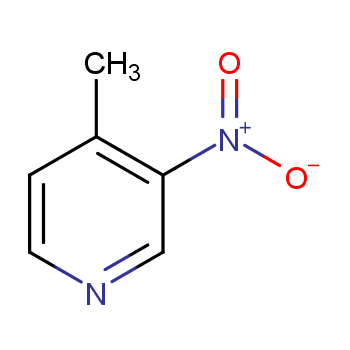
本文将介绍高效合成3-硝基-4-甲基吡啶的方法,希望能够为3-硝基-4-甲基吡啶的合成提供新思路和新方法。
背景:3-硝基-4-甲基吡啶(1)是化工及药物中间体。可由4-甲基吡啶通过N2O5硝化制得,也可由3-硝基-4-甲基吡啶-2-甲酸在210℃高温脱羧制得。
合成:
从常见的工业品2-氨基-4-甲基吡啶(2)作为起始原料,经过混酸硝化制备得到3-和5-硝基取代的混合物(3)。接着,将2-氨基基团重氮化为2-羟基,然后使用POCl3和PCl5作为卤代试剂进行氯化得到5。最后,在酸性介质中,在苯甲酸的存在下,利用铜粉进行脱氯反应,最终得到产物1,总收率为60%。具体实验操作如下:
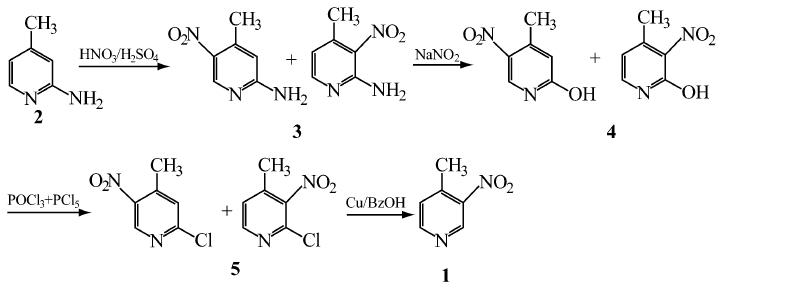
(1)2-氨基-4-甲基-3(5)-硝基吡啶(3)
将950克(9.5摩尔)浓硫酸加入到1000毫升的四口烧瓶中,并在搅拌的情况下分批加入2(108克,1.0摩尔)。在10℃以下滴加108克(1.1摩尔)浓硝酸,滴加完毕后,在室温下反应12小时,然后在95℃下反应2小时。冷却至室温后,将混合物转移到1000毫升的冰水中,使用浓氨水调节至pH7,然后进行过滤以得到3,该产物可直接用于下一步的反应。
(2)2-羟基-4-甲基-3(5)-硝基吡啶(4)
于3000ml四颈烧瓶中加入如上所得3、水(2L)及浓硫酸(260g,2.6mol),10℃以下滴加NaNO2(83g,1.2mol)的水(240ml)溶液,滴毕,保温反应1h,10℃以下过滤,滤饼于80℃干燥,得淡黄色固体4[131g,85%(以2计)]。
(3)2-氯-4-甲基-3(5)-硝基吡啶(5)
于1000ml四颈烧瓶中加入4(108g,0.7mol)、PCl5(21g,0.1mol)和POCl3(200g,1.3mol),回流反应6h。常压回收POCl3后将反应混合物缓慢倒入冰水(700ml)中,静置,分出有机层,水相用CHCl3(100ml×4)萃取,合并有机相和萃取液,用无水硫酸钠干燥。过滤后常压回收溶剂,残液减压蒸馏,收集110~120℃/4.0kPa馏分,得淡黄色液体5(115g,95%)。产品通过GC-MS测定,在10.7和11.1min处出现两个峰,含量分别为38.6%和61.3%,其m/z均为172(M+)。
(4)4-甲基-3-硝基吡啶(1)
于1000ml四颈烧瓶中加入5(69g,0.4mol)和苯甲酸(122g,1.0mol),在150℃下分批加入活化铜粉(64g,1.0mol),加毕保温反应30min,冷却后加入20%Na2CO3水溶液(400ml),水蒸气蒸馏,馏出液用氯仿(100ml×3)萃取,用无水硫酸钠干燥。过滤后常压回收溶剂,残液减压蒸馏,收集145~155℃/4.0kPa馏分,得淡黄色液体1(41g,74%)。
参考文献:
[1]孙楠,莫卫民,胡宝祥等.4-甲基-3-硝基吡啶的合成[J].中国医药工业杂志,2003,(12):7-8.


 1
1