提问
问答首页
问题分类
文章
视频
消息管理
站内通知
我的
盖德化工网
盖德化工网
>
盖德问答
>
Aspen中用UNI...
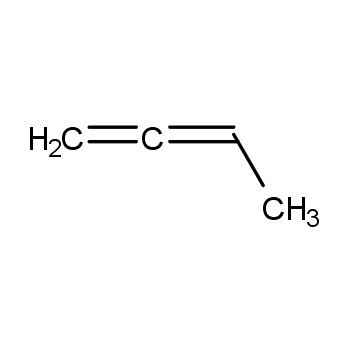
Aspen中用UNIFAC模拟1,2-丁二烯,丙二烯等出现的问题?
Aspen中用UNIFAC模拟1,2-丁二烯,丙二烯等,即一个碳原子上有两个双键时,会出现如下的提示: 请问怎么解决?
0评论
+
关注
共10个回答
丧偶,工艺专业主任
2018-08-04回答
a+中unifac定义了大多数官能团,但没有定义所有的基团。你说的这两个物质中的二烯键(=c=)是unifac没有定义的。unifac只能定义chm=chn,m,n=0~3。所以间隔的二烯是可以用unifac估算的,你说的这两个二烯是连续的,unifac没办法定义。就出error了。
2
评论 0
举报
standbyme,销售
2018-08-04回答
unifac模拟1,2-丁二烯,丙二烯等的什么物性?
2
评论 0
举报
爱你的我会宠,工艺专业主任
2018-08-04回答
a+中用unifac方法计算该体系的方法: a+中新定义基团c3h4和c3h3,bondi基团定义c3h4, 107 1个、104 2个;c3h3, 107 1个、104 1个、105 1个;然后就可以利用unifac定义丙二烯和1,2-丁二烯,其中丙二烯含c3h4 1个,1,2-丁二烯含c3h3 1个、-ch3 1个。 最后利用heater模型求算泡点压力。
展开
9
评论 0
举报
清风微微起,工艺专业主任
2018-08-04回答
无论什么都不可以,用unifac算一个碳原子上有两个双键的物质就报错。 举个例子,就50%的1,2-丁二烯和50%的丙二烯,求50度下的泡点压力。 怎么弄?
6
评论 0
举报
狗樣,工艺专业主任
2018-08-04回答
为什么要回复之后才能看到呢
6
评论 0
举报
专属迷妹,改性塑料销售
2018-08-04回答
像类似于这种a+中没有定义的基团,可以自己在a+中定义(也就是利用bondi等方法定义设定的unifac基团),求出它的r\q,代入进行计算。 不知道lty讲的pro2的方法是什么方法?
展开
2
评论 0
举报
殉情,仪表工
2018-08-04回答
楼主这是在什么条件下的物系啊?纯学术探讨还是实际情况?1,2-丁二烯这物质也能稳定存在?
9
评论 0
举报
野冢.,Sales
2018-08-04回答
proii直接就能计算。
8
评论 0
举报
情初°初晴°,给排水工程师
2018-08-04回答
这两个组分在proii中有不太严格的集团拆分法,因此proii默认是可以计算的,按照proii的方法在aspen中指定对应的集团,aspen就可以计算了,两者计算结果相差不太多。
展开
7
评论 0
举报
书生,工艺专业主任
2018-08-04回答
刚用a+试了一下,a+利用unifac计算50%的1,2-丁二烯和50%的丙二烯在50度下的泡点压力,8.567bar。a+利用pr计算50%的1,2-丁二烯和50%的丙二烯在50度下的泡点压力,8.491bar。结果相近。
展开
6
评论 0
举报
上一条:
灰水加热器换热效果差,怎么处理?
下一条:
专业考试天大版6-32有误?
1,2-丁二烯
CAS No:590-19-2
国内供应商(24家)
湖北万得化工有限公司
VIP
14年
备案
联系电话:
15377098680 座机:027-59210159
产品名称:
1,2-丁二烯
价格:
1元 /千克
主营产品:
N-乙烯基己内酰胺(2235-00-9)、4,4'-亚甲基双(2,6-二甲苯酚)、多库酯钠、二苄基甲苯、
在线询盘
洽谈
湖北恒景瑞化工有限公司
VIP
7年
备案
联系电话:
产品名称:
1,2-丁二烯
价格:
电询
主营产品:
邻碘苯甲酸、阿奇霉素、二苯硫醚、2-氯苯并噻唑、
在线询盘
洽谈
湖北万得化工有限公司
VIP
6年
备案
联系电话:
027-59210159
产品名称:
1,2-丁二烯
价格:
1元 /千克
主营产品:
万得优势产品 乙酸锶 欢迎询价、万得主打产品 1,3,5-三嗪-2,4,6-三胺氢溴酸盐; 三聚氰胺氢溴酸盐 欢迎询价、万得优势产品 二烷基萘 欢迎询价、万得优势产品1-(2-羟乙基)-2-咪唑啉酮 欢迎询价、
在线询盘
洽谈
湖北实顺生物科技有限公司
VIP
6年
备案
联系电话:
15172526927 座机:027-59223025
产品名称:
1,2-丁二烯
价格:
1元 /千克
主营产品:
苄基甲苯、辛基膦酸(4724-48-5)、4-氯 甲基苯 乙烯(1592-20-7)、对甲苯磺酸(104-15-4)、
在线询盘
洽谈
湖北广奥生物科技有限公司
VIP
11年
备案
联系电话:
18162699091 座机:027-59223056
产品名称:
1,2-丁二烯
价格:
1元 /千克
主营产品:
二甲胺基丙胺二异丙醇 DPA、硬脂酸铁(555-36-2)、苄氧基胺盐酸盐2687-43-6、索马鲁肽、
在线询盘
洽谈
查看更多供应商 >
1,2-丁二烯相关回答
Aspen中用UNIFAC模拟1,2-丁二烯,丙二烯等出现的问题?
10个回答
丁二烯抽提装置中1,2-丁二烯是如何去除的?
7个回答
如何制备2-三甲基硅氧基-1,3-丁二烯?
1个回答
液体1,2-聚丁二烯求购?
1个回答
2-乙炔基甲苯与1,3-丁二烯反应方程式?
1个回答
您可能感兴趣的问答
三价铬溶液颜色问题?
15个回答
能否用离心代替旋蒸去除乙醇?
2个回答
想请教下靛蓝染料在紫外分光光度计下吸收的问题?
4个回答
硫酸钙结垢,用什么清洗掉?
4个回答
氰基取代苯环上的卤素的反应条件?
0个回答
请填写举报原因
选择举报原因
垃圾广告
有害信息
文不对题
涉嫌侵权信息
其他
增加悬赏
剩余能量值
能量值
提示
提示信息
确定
取消
消息提示