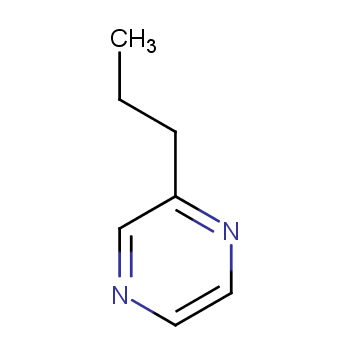
2-丙基吡嗪是一种常用的医药合成中间体,可以通过2-氯吡嗪和丙基溴化锌在乙酸钯和L10·HBF4的催化下反应得到。

首先,在装有磁性搅拌棒和特氟龙隔膜的烘箱中干燥的螺帽试管中加入乙酸钯(2.3mg,0.01mmol)和L10·HBF4(8.3mg,0.01mmol),然后加入2-氯吡嗪。将试管抽空并用氩气反吹,重复此过程3次。将反应混合物冷却至0℃,然后滴加丙基溴化锌的THF溶液(2.0mL,0.64M的THF溶液,1.30mmol)。除去冰浴后,在室温下搅拌2小时,然后用水(5mL)淬灭反应,并用乙酸乙酯(3×10mL)进行萃取。将合并的有机相经过MgSO4干燥,真空浓缩,并通过Biotage Isolera纯化,得到淡黄色油状物2-丙基吡嗪(85mg,70%收率)。(注意:产品易挥发,长时间在高真空下放置可能导致收率降低。)
将8.0mL 2-氯吡嗪、1.58g乙酰丙酮铁和100mL THF(四氢呋喃)加入搅拌的圆底烧瓶中。在氮气下搅拌,得到红色溶液。将烧瓶放入冰水浴中冷却十分钟。然后开始向烧瓶中加入49mL氯化正丙基镁,得到深紫色溶液。1.5小时后,在十分钟内再加入10mL氯化正丙基镁。再过20分钟后,再加入5mL氯化正丙基镁,搅拌约30分钟后,在7分钟内加入22mL饱和NH4Cl水溶液。加入另外的7mLNH4Cl后,停止搅拌并将混合物在室温下在氮气下静置过夜。加入125mLEtOAc和450mL水后,将烧瓶的内容物通过聚丙烯过滤并倒入分液漏斗中,分离各相,并将水相用125mL的EtOAc再萃取两次。合并的有机相通过硅藻土过滤,随后通过旋转蒸发(200mbar,40℃)浓缩,在短程蒸馏之后,将馏出物通过Vigreux柱蒸馏(200mbar,90-110℃),得到9.0g(82.4%收率)的2-丙基吡嗪。
[1] Highly Selective Palladium-Catalyzed Cross-Coupling of Secondary Alkylzinc Reagents with Heteroaryl Halides
[2] WO2010107969


 1
1