本文旨在探讨关于1-溴-2,3-二氯苯的电子和结构方面的研究进展及相关问题,通过对该领域的深入研究,我们希望能够为1-溴-2,3-二氯苯的相关应用提供有益的指导和启示。
简述:1-溴-2,3-二氯苯(BDCB),英文名称:1-Bromo-2,3-dichlorobenzene,CAS:56961-77-4,分子式:C6H3BrCl2,外观与性状:白色至浅黄色结晶。1-溴-2,3-二氯苯是一种用于制备富纹状体蛋白酪氨酸磷酸酶选择性非肽抑制剂的试剂。
电子和结构方面的综合研究:
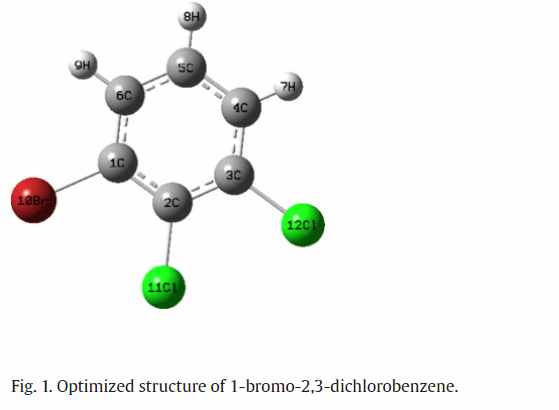
(i)通过FT-IR和FT-Raman光谱,13C和1H NMR以及UV/vis对BDCB(图1)进行结构研究。(ii)应用密度泛函理论(DFT)计算了1-溴-2,3-二氯苯的分子几何形状和振动谱。(iii)采用积分方程形式-极化连续体模型(IEF-PCM)介绍了溶剂对13C和1H NMR数据的影响。(iv) HOMO-LUMO和NBO分析已被用于提供分子内电荷转移的更多信息。理解溶剂效应的一个关键方法是溶剂引起的溶质电子跃迁的变化,通常称为溶剂致变色。M Arivazhagan等人报道了不同溶剂对化合物电子谱和核磁共振谱的影响。具体如下:
(1)实验细节
BDCB 的纯样品购自英国兰卡斯特化学公司,并用于光谱测量。使用 BRUKER IFS-66V 型 FT-IR 光谱仪在 4000–400 cm?1 区域记录该化合物的 FT-IR 光谱。化合物的 FT-拉曼光谱记录在配有 FRA-106 FT-拉曼附件的 BRUKER IFS-66V 型干涉仪上,在斯托克斯区域 (3500–50 cm?1),使用 Nd:YAG 激光器在 200 mV 功率下连续运行,1064 nm 激发。校准后的波数预计准确度在±1 cm?1 以内。
(2)计算细节
使用 GAUSSIAN 09W ,通过 HF/6-311++G(d,p) 和 DFT 6-311++G(d,p) 方法优化基态化合物的气相几何形状程序。对于这种优化的几何形状,在相同的理论水平上分析计算振动频率,以确保它是真正的局部最小值。
在NMR计算中,首先采用积分方程形式化极化连续介质模型(IEFPCM)方法在CDCl3(ε = 4.9)中在6-311++G(d,p)基础水平上获得了BDCB的优化分子几何结构。同样,通过相同的方法在DMSO(ε = 46.7)中获得了优化的BDCB分子几何结构。然后,使用规范不变原子轨道 (GIAO) 方法计算溶剂中 B3LYP/6-311++G(d,p) 水平下 BDCB 的 1H 和 13C NMR 化学位移。
此外,使用 B3LYP 方法和 6-311++G(d,p) 基组计算了 BDCB 的 HOMO 和 LUMO 能量值以及能隙。此外,通过使用 B3LYP/6-311++G(d,p) 水平的优化结构以 3D 形式绘制 BDCB 的分子静电势 (MEP)。
经过基态几何优化后,在 PBE1PBE/6-311+G(d,p) 水平上计算了化合物的激发光谱。基组和函数的选择清楚地表明:(a) 偏振函数的加入并没有给激发能的计算带来任何显着的改进;(b) 添加额外的偏振函数仅使 λmax 增加 1 nm,表明不需要第二组偏振函数;(c) 纯 DFT 泛函提供的激发能量太小,而使用 25% 泛函(尤其是 PBE1PBE)可以达到最佳精度。由于溶剂效应对化合物的吸收光谱起着重要作用,在激发能计算中选择处理溶剂效应的积分方程形式化极化连续介质模型(IEFPCM)。在 IEFPCM 中,将问题分为位于空腔内的溶质部分和表示为无结构材料的溶剂部分,以介电常数和其他宏观参数为特征。用B3LYP方法计算了化合物在同一基集水平下不同温度下的热力学性质。
(3)拉曼强度的预测
随后使用源自拉曼散射基本理论的以下关系将拉曼活动转换为相对拉曼强度 (Ii):
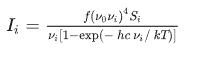
其中 ν0 是激光激发波数,单位为 cm?1(在本工作中,我们使用激发波数 ν0 = 9398.5 cm?1,对应于 Nd:YAG 激光器的 1064 nm 波长),νi 是振动波数第 i 个简正模式 (cm?1) 的拉曼散射活度,而 Si 是简正模式 νi 的拉曼散射活度。f(是等于 10?12 的常数)是为所有峰值强度适当选择的通用归一化因子。h、k、c 和 T 分别是普朗克常数和玻尔兹曼常数、光速和开尔文温度。
(4)结论:
本研究为理解1-溴-2,3-二氯苯(BDCB)的电子和结构方面提供了一个完整的方法。结果表明,考虑溶剂效应时,6-311+G(d,p)基的PBE1PBE杂化泛函能提供可靠的λmax。
NBO 分析显示,研究的化合物具有电子转移的结构特征。前线分子轨道(HOMO-LUMO)负责电子极化和电子转移性质。通过将电子密度映射到静电势表面(MESP)来识别反应性位点。此外,使用规范不变原子轨道(GIAO)方法计算了 13C 和 1H。由于第一超极化率较高(比尿素(0.37289 × 10^-30 esu)高 5.7 倍),1-溴-2,3-二氯苯可以用作良好的非线性光学材料。最后,值得一提的是,溶剂导致吸收峰值从气相向红移,并且从极性较小的溶剂向极性较大的溶剂过渡 S0 → S1 会略微向蓝移。
参考文献:
[1]Arivazhagan M, Muniappan P, Meenakshi R, et al. PCM/TD-DFT analysis of 1-bromo-2, 3-dichlorobenzene–a combined study of experimental (FT-IR and FT-Raman) and theoretical calculations[J]. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2013, 105: 497-508.


 1
1