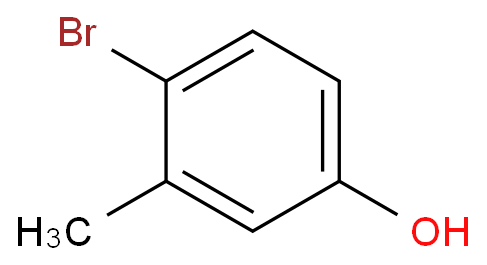
4-甲基-3-溴苯酚的合成方法及其改进是一个引人注目的研究领域,通过创新的方法和改进的技术,我们可以有效地合成这种化合物。
背景:4-甲基-3-溴苯酚(1)是一种重要的有机合成中间体,可用于合成其醚化物、芳氧羧酸类衍生物、苄醇类化合物等。文献报道,1主要是以4-硝基甲苯(5)为起始原料,经溴化、还原、重氮化和重氮盐的水解4步反应合成。
合成改进:
任志强等人对4-甲基-3-溴苯酚的合成进行了改进,简化了溴化反应的后处理步骤,解决了还原反应的后处理中Sn(OH)2胶体沉淀难以破坏的问题,以及改进了重氮盐水解反应的操作方法。根据改进的方法,通过4-硝基甲苯为起始原料,经过4步反应合成目标化合物,总收率为53%。具体步骤如下:
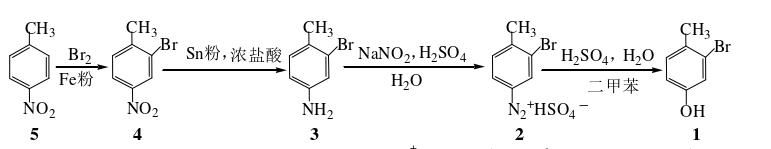
(1)4-硝基-2-溴甲苯(4)的合成
在250毫升的四颈瓶中,首先加入4-硝基甲苯(5)34.3克(0.250摩尔)和0.5克铁粉,然后加热并控温在75~80℃,在电动搅拌下,30分钟内滴加15.3毫升溴(47.7克,0.299摩尔)。溴加完后继续保温反应1.5小时。将反应混合物倒入250毫升冷水中,会出现大量淡黄色晶体,并有Fe(OH)3胶体沉淀出现,在搅拌下滴加浓盐酸至Fe(OH)3沉淀消失,共加入浓盐酸8毫升。随后进行减压抽滤,水洗,干燥,得到4的粗品。用冰醋酸进行重结晶,得到淡黄色针状晶体4,重48.0克,收率为88.9%。熔点为74~75℃。
(2)4-甲基-3-溴苯胺(3)的合成
在250 mL四颈瓶中,加入4 21.6 g(0.100 mol)和锡粉26.1 g(0.220 mol),在电动搅拌、冰水浴冷却下慢慢滴加浓盐酸56 mL, 保持温度在60 ℃以下。加完浓盐酸后撤去冰水浴,加热回流2 h, TLC监测至反应完全(硅胶GF-254;展开剂:乙酸乙酯∶石油醚 =1∶4)。待反应液冷至80 ℃左右,在电动搅拌下15 min内滴入 40 %的氢氧化钠溶液45 mL,此时出现大量灰黑色沉淀。冷却后减压抽滤,滤液用乙醚萃取(25mL×2),用饱和食盐水25mL洗涤 1次,无水硫酸钠干燥,常压蒸除乙醚后减压蒸馏,收集146~148 ℃/4.6×103 Pa(35 mmHg)馏分,得淡黄色油状液体3,重14.5 g,收率78.0 %。
(3)4-甲基-3-溴苯酚(1)的合成
在100 mL三颈瓶中,加入3 5.6 g(0.030 mol)及由浓硫酸6 mL 和水15 mL配成的热的稀硫酸,磁力搅拌3 min后用冰水浴冷至 15 ℃,向反应液中加入碎冰10 g。当反应液混合物降至5 ℃时,在搅拌下滴加由亚硝酸钠2.42 g(0.035 mol)和水7 mL配成的溶液,并保持反应液温度在5 ℃以下,约15 min滴完,然后继续搅拌5 min。加入冷水6 mL和尿素0.24 g,搅拌10 min。由此制得重氮盐2的溶液,冰水浴中冷却备用。在250 mL三颈瓶中加入二甲苯50 mL,在回流状态下分批滴加2的溶液,约1 h加完,然后继续回流5 min。冷却后分出有机相,并用二甲苯萃取水相(25 mL×2)。合并有机相,加入5 %氢氧化钠溶液50 mL,充分振摇后分出水相。加入10 %盐酸25 mL至强酸性,用乙醚萃取(25 mL×2),饱和食盐水10 mL洗涤,无水硫酸钠干燥。常压蒸除乙醚后,得1的粗产品,为橙红色油状物。用石油醚重结晶,活性炭脱色,得无色针状晶体1,重4.3 g,收 率77 %。m.p.55~56 ℃。
参考文献:
[1]任志强,秦丙昌.4-甲基-3-溴苯酚合成的改进[J].广东化工,2014,41(08):33+36.


 1
1