本文旨在探讨关于5-氟-2-甲基苯甲腈理论光谱和第二谐波产生的研究进展及相关问题,希望能够为5-氟-2-甲基苯甲腈的相关应用提供有益的指导和启示。
简述:苯甲腈是母体分子氨与甲苯的子分子,其随特定比例的结果而发生巨大变化。香料、化妆品、类固醇、脱色剂用芳香醇、脂肪酸溶剂、碳氢化合物和油类与苯甲腈一起用作化学剂。近年来,研究人员将苯甲腈作为许多衍生物中最优良的溶剂和多功能前体化学中间体。5-氟-2-甲基苯甲腈(5F2MLBN) 中的苯环具有三个取代基,使得氟基和甲基相对于氰基分别处于间位和邻位。甲基和氰基通常被称为芳香环系统中的供电子取代基。苯甲腈和芳香环中氰基的结合产生了氮原子的迁移。
理论光谱和第二谐波产生的研究:
Kumar A A P等人记录了5F2MLBN的FT-IR(400-4000 cm-1)和FT-拉曼光谱(50-3500 cm-1)。确定了具有三个较高基组的密度泛函HF方法,确定了5F2MLBN在基态下的分子几何结构,谐波振动频率和结合特征。最后得到5-氟-2-甲基苯甲腈的光谱具有良好的互补性,同时与计算结果进行了比较,以模拟红外光谱和拉曼光谱。
1. 实验部分:
(1)实验细节
光谱分析的目的,傅里叶变换红外(FTIR)和傅里叶变换拉曼(FT Raman)光谱分别在常温下记录在400-4000cm-1和50-3500cm-1的区域。红外光谱使用8400S Bruker、Alpha T和德国红外分光光度计记录,扫描速度为30 cm-1 min-1。FT Raman光谱使用Nd: YAG激光器的1064nm波长在EZRaman、Enwaveoptronics和美国IFS 66 V光谱仪上记录。
(2)计算细节
Gaussian 09W 软件包已用于预测5F2MLBN的整体振动分配和优化原始版本的几何参数。完整的几何参数是从Becke-Lee-Yang-Parr混合(B3LYP)方法中得出的,通过在Intel Core i3 3.3GHz处理器个人计算机上应用从头算B3LYP混合方法。通过引入缩放因子校正的振动波数。缩放因子值分别为0.9556和0.9959,对应(d,p)和cc-pvdz 。通过使用VEDA.4.0.Software的缩放量子力学程序计算和解释了总能量分布(TED),并通过其TED计算了振动模式。通过能量的二阶导数计算了FT-IR、拉曼频率,并通过Gauss sum程序绘制了强度图。计算了非线性光学(NLO)特性,如偶极矩(μ)、极化率(α)、极化率的各向异性(α0)和第一超极化率(β0),以了解5F2MLBN的倍频SHG行为,通过Gaussian09 DFT计算了热力学参数。
2. 结果:
(1)分子几何优化
5F2MLBN化合物的优化结构已经被说明(见图1)。通过B3LYP功能和标准的三个基组计算得出的稳定最小能量。通过文献调查,5F2MBN的优化结构和平衡参数数据在先前的报告中并不存在。与理论值相比,微波数据稍微偏小,优化原子长度的理论值在气相中处于正确位置的独立的目标化合物。两种不同基组的预测几何参数几乎相似。理论值与微波数据有很好的一致性。苯环有六个碳原子和氢原子,碳原子具有相同的长度和角度,氢原子有一些变化。由于苯环中的氢原子作为价电子分布的扭曲,分子在不同的化学和物理性质上发生了改变。最近的分子相互作用表明苯环角度的变化。


该分子有七个碳键,八个碳-碳键,六个碳-氢键,一个碳-氮键和一个碳-氟键。5F2MLBN没有晶体结构,苯环在碳-碳原子键长的替位位置(≈1.40?)处显得有点朦胧,比环中被氟取代的碳键(C2-F9)(≈1.35?)长。扭曲表明环上的取代物可以影响碳原子的杂化和键长。与苯环相比,碳-碳键长增加,显示出替代原子之间的角度稍微改变了六角结构的角度。从这个结果可以看出,苯环的碳键长度几乎相同,偏差应该在0.005 ?左右,这与早期的报告相符。所有基本集计算得出的C≡N键长为1.15?,早期结果中对苯腈观察到了1.15 ?。所有结果都有很好的相关性,最小偏差水平为0.02?,这个结果比碳单键和氟基的1.35 ?短。最后,计算了碳氟键的长度,并与观察值合并。键之间的角度是由一个原子预测的,与微波数据进行比较得出了相应的结果。
(2)光学特性
超极化率是由分子的结构、键合和振动决定的。苯甲腈的偶极矩(μ)和超极化率很高。在最近的分子中已经计算出增强的超极化率值,这是由于苯甲腈的取代基。键和振动结果证实了5F2MLBN分子的超极化性扩大。在电话、信号传输和光缆等领域,NLO增强了调频、光变、光控制和光逻辑电路等技术发展中的功能。5F2MLBN的第一超极化率(β)、极化率(α)和偏振率的各向异性(α)是用DFT计算的,可以分别用方程(1)、(2)、(3)来计算。表4列出了上述参数的数值。

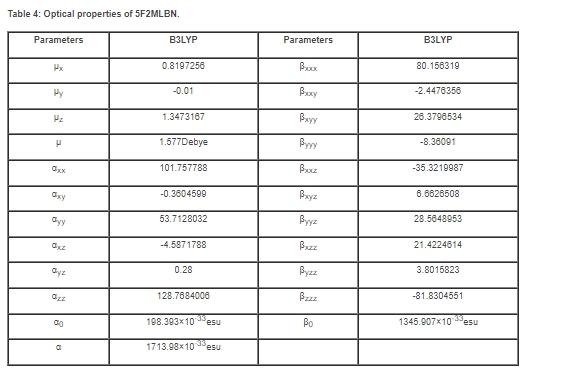
α和β的计算值分别为1713.98×10?33esu和1345.907×10?33esu。理想分子尿素被用来确定比较目的。该化合物的μ和β值分别为1.577D和1345.907×10-33esu。该化合物的μ大约比尿素大1.15倍,β大约比尿素大3.62倍(尿素的μ和β值分别为1.3732 Debye和0.3728 ×10-30esu,采用相同的方法)。最近的研究已确认该化合物具有稳定的非线性光学应用潜力,如频率加倍和通信。
(3)结论
从DFT方法中分三基集,计算出几何参数并优化结构。该研究首次研究了实验性 FT-IR、FTR 光谱研究,并使用 B3LYP 方法解释了 TED% 的振动分配。采用计算方法预测了理论光谱,并与实验结果吻合较好。该化合物的μ值和β值分别是尿素的1.15倍和3.62倍。这些特性表明,5F2MLBN具有良好的化学稳定性,生物活性和光学应用,可帮助未来的研究人员和创新思想家。
参考文献:
[1]Kumar A A P, Raman R G. Theoretical Spectroscopic and Second Harmonic Generations Studies of 5–Fluoro-2-Methyl Benzonitrile[J]. Oriental Journal of Chemistry, 2017, 33(5): 2412-2420.


 1
1