各位,最近在检测
罗库溴铵注射液稳定性,所用色谱柱为赛默飞正向硅胶柱(250*4.6mm,3μm),流动相为4.53g/L四
甲基氢氧化铵五水合物的水溶液:乙腈=1:9,混匀后超声3-5min。
色谱柱平衡约4h基线平稳后便进样,顺序依次为空白溶液(即流动相)、分离度溶液(罗库溴铵
标准品和杂质C标准品的混合溶液)、含量对照品溶液(罗库溴铵标准品溶液,要走五针)和含量重复对照品溶液(罗库溴铵标准品溶液)。
第一针空白完全正常;第二针分离度溶液中罗库溴铵峰和杂质C峰出峰时间和峰面积均正常,RT分别为11min和13min,但峰高较之前低,且在图谱后半段有一个小鼓包,再往后基线上抬(图1);第三针含量对照品溶液第一个出峰在14min,且随后跟着一个大鼓包(图2),查看其压力时发现在鼓包的位置压力突然抖动了一下(图3);第四针含量对照品溶液走到一小半基线突然往下掉,感觉也没有峰出来(图4);第五针含量对照品溶液则完全没有峰(图5) 。后来我们将分离度溶液再次进样(图6),按照原有的积分方法并不能积出,之后我们发现在2.8min处有个小峰(图7),这与我们之前做罗库溴铵时出现的情况类似(RT=4min左右,峰型类似)。而之前我们将原因归结为流动相混匀时的操作不当,现在看来原因可能并非如此。
各位谁之前遇到过这种情况,或者对此比较熟悉的,还希望帮个忙分析一下,不胜感感激!

图1 第二针 分离度溶液.jpg

图2 第三针 含量对照品溶液.jpg

图3 异常压力图.jpg
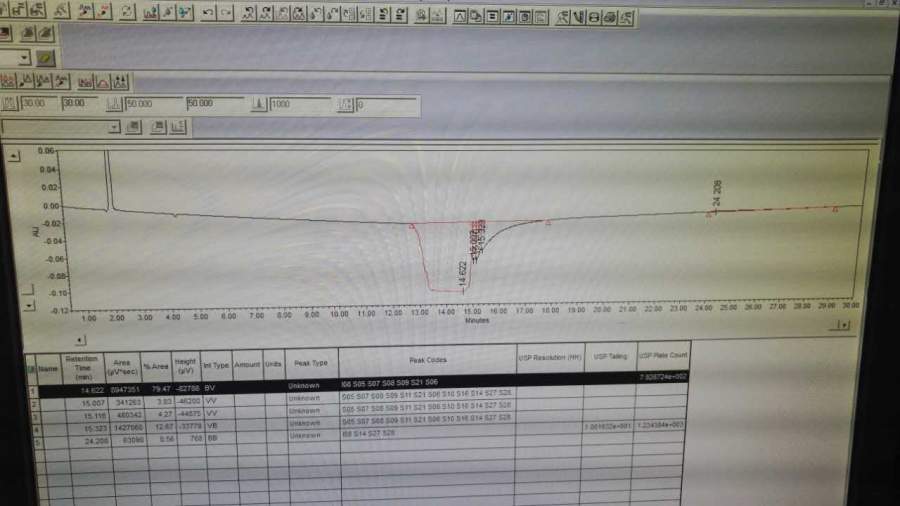
图4 第四针 含量对照品溶液.jpg

图5 第五针 含量对照品溶液.jpg

图6 再次进样的分离度溶液.jpg
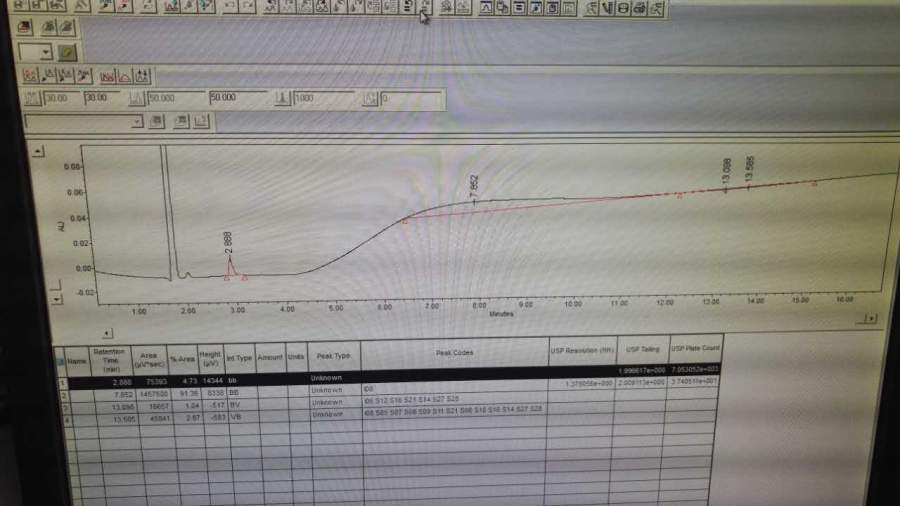
图7 再次进样的分离度溶液(手动拉峰).jpg

正常压力图.jpg