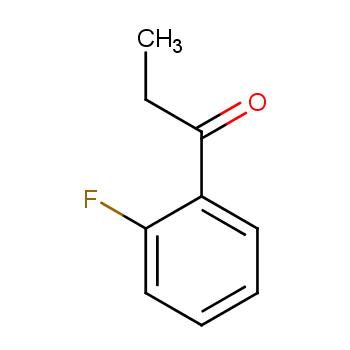
合成邻氟苯丙酮是合成化学中的重要课题。本文旨在探讨有效的方法来合成邻氟苯丙酮,以满足其在相关领域中的应用需求。
背景:邻氟苯丙酮(1)是合成新型抗瘫药和肌肉松弛剂2-甲基-1-(2-氟苯基)-3-哌啶基-1-丙酮的关键中间体,该药在国外已用于临床。
合成:
1. 邻氟甲苯(2)的合成
使用165毫升浓盐酸,在搅拌下缓慢滴加107克邻甲苯胺(1摩尔),加热搅拌至固体完全溶解后,冷却至0℃。然后滴加1.1摩尔亚硝酸钠与100毫升水的溶液,温度不超过4℃,使用碘化钾-淀粉试纸检测终点,过量的亚硝酸钠用氨基磺酸除去。同时制备1.3摩尔的氟硼酸(硼酸加氟化氢),冷却至0℃(留50毫升作为洗涤用),倒入制备好的、冰冷的上述重氮盐中,在搅拌下2分钟后抽滤,滤饼分别用冷的氟硼酸50毫升、95%乙醇溶液、乙醚(50毫升×2)洗涤抽干后凉干,真空干燥(不加热)。进行热分解,馏出物用10%氢氧化钠溶液(20毫升×3)、水(2毫升×2)洗涤,无水碳酸钠干燥,常压精馏收集113~115℃产品,得到无色液体2(71.5克,65%)。
2. 邻氟苯甲醛(3)的合成
110 g(1 mol)化合物2,0.5 g过氧化苯甲酰,加热至110℃回流时在光照下通入氯气,调节热源以保持微沸,6 h后bp为180℃,增重50 g,继续通氯气,2 h后升温到190℃(增重70 g)时,停止通氯气。通入氮气除去氯化氢,减压精馏,于82~86℃/5.3 kPa蒸出“一氯化物”,剩下的“二氯化物”和“三氯化物”不分离,直接水解。
上述“二氯化物”,氧化锌2 g,水600 mL,回流1 h后,利用水份分离器分出比水重的乳白色油状物,用CH2Cl2(50 mL×2)萃取,并用10%的碳酸钠溶液洗涤,无水硫酸镁干燥。常压精馏收集110~115℃/101.3 kPa产物,得无色液体3(90.5 g,80%)。“二氯化物”水解后的水溶液和洗涤后的水溶液加10%氢氧化钠溶液至pH 12,滤出不溶物,滤液加入盐酸至酸性,抽滤、干燥得白色固体5(10 g,8%),mp 124~126℃。
3. 邻氟苯丙醇(4)的合成
2.6 g(0.11 mol)镁屑和5 mL无水乙醚,搅拌下滴入溴乙烷12 g(0.1 mol)和20 mL无水乙醚的混合液,保持溶液微沸,加完后回流1 h,冷却和搅拌下滴入12.4 g(0.1 mol)化合物3和20 mL无水乙醚的混合液,加完后回流15 min,冷却和搅拌下滴入15 g氯化铵的饱和溶液以分解产物,加入50 mL 50%盐酸溶液使固体全部溶解。分出醚层,水层用乙醚(50 mL×2)萃取,合并乙醚层,用碳酸钠溶液、水洗涤,无水硫酸镁干燥, 减压精馏,收集95~99℃/2.5 kPa产品,得无色液体4(13.7 g,90.4%)。含量为99.2%(HPLC)。
4. 邻氟苯丙酮(1)的合成
9 g(0.058 mol)化合物4滴入80 mL水,3 mL硫酸和8 g(0.08 mol)铬酸混合液中,温度保持45℃左右,反应3 h后加入500 mL水,分出油层,水层用CH2Cl2(50 mL×3)萃取,合并油状物用饱和碳酸氢钠溶液(50 mL×3)洗涤,无水硫酸镁干燥,蒸去CH2Cl2,得目标化合物1(8 g,92.6%)。
参考文献:
[1]陈红飙,林原斌. 邻氟苯丙酮的合成 [J]. 合成化学, 2002, (02): 146-150.
[2]陈红飙,林原斌. 邻氟苯丙酮的合成研究 [J]. 中国药物化学杂志, 2002, (01): 33-36.


 1
1