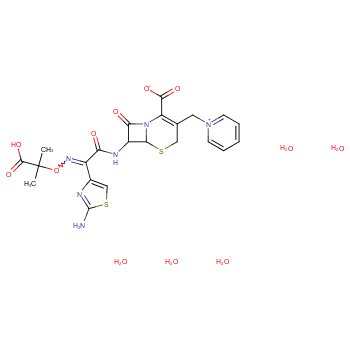
头孢他啶侧链酸活性硫酯的合成方法研究不仅拓宽了头孢类药物的合成途径,还为药物领域的进一步探索提供了新的思路。这项研究对于开发新型抗菌药物具有重要意义,并可能在临床上取得突破性的应用。
简介:头孢他啶侧链酸活性硫酯是合成头孢他啶的重要中间体,它的品质和收率直接影响所合成药物头孢他啶的质量好坏和成本。因此,开发成本低、收率高、操作简单的头孢他啶侧链酸活性硫酯合成工艺,对我国医药工业的发展具有重要意义。
1.方法一
头孢他啶侧链酸与DM缩合生成头孢他啶侧链酸活性硫酯。头孢他啶侧链酸在碱性条件下与DM缩合生成头孢他啶侧链酸活性硫酯时,用价格低廉的亚磷酸三乙酯代替价格昂贵的三苯基膦, 通过设计正交实验优选出的较佳反应条件为:固定头孢他啶侧链酸19.8 g,反应物料n(头孢他啶侧链酸) ∶n(DM)∶n(亚磷酸三乙酯)∶n(三乙胺)=1∶1.2∶1.2∶1.2,反应溶剂乙腈150 mL,于5℃反应5 h,收率86.8%。

具体步骤为:向装有冷凝管和机械搅拌装置的四口瓶中依次加入三苯基膦(20.5 g、0.078 mol)、精制的DM(25 g、0.078 mol)、三乙胺(6.03 g、0.06 mol)于75 mL甲苯和75 mL二氯甲烷的混合液中,搅拌1 h.在2 h内分批加入头孢他啶侧链酸(19.8 g、0.06 mol),控制温度不超过5 ℃,加完后搅拌4 h,将反应液冷冻,抽滤, 分别用甲苯50 mL和乙醚30 mL洗涤,60 ℃下真空干燥,得到浅黄色固体16.6 g,收率为75%.产品mp为134 ℃~136 ℃,Rf=0.42(硅胶板,展开剂PE∶AcOEt=3∶1)。
2. 方法二
以去甲氨噻肟乙酸乙酯(DMAT3)为原料,与α-溴代异丁酸叔丁酯进行醚化反应,生成头孢他啶侧链酸酯(4),再将化合物4进行选择性水解得到头孢他啶侧链酸(1),化合物1与二硫代二苯并噻唑 (DM)进行S-酰化得到目标产物——头孢他啶活性硫酯(2)。即以乙酰乙酸乙酯为起始原料,经肟化、溴化、成环3步反应合成去甲氨噻肟乙酸乙酯[2-氨基-α-羟亚氨基-4-噻唑乙酸乙酯];再经醚化、水解、酰化反应合成其活性硫酯-{(Z) -2-氨基-α-[[2-叔丁氧基)-1,1-二甲基-2-氧代乙氧基]亚氨基-4-噻唑乙酸2’-苯并噻唑基硫酯。

3.方法三
三颈瓶中依次加入侧链酸30g DM58.5g Ph3P 46.3g二氯甲烷460ml升温 至27℃,加入三乙胺10.4m1,35℃下搅拌反应2.5小时。降温度至0~5℃ ?搅拌30分钟,过滤,用0~5℃二氯甲烷洗三次,25~30℃干燥5小时。头孢他啶活性硫酯的质量很好,完全能够达到头孢他啶生产过程中的质量要求。
4.方法四
在醇水溶剂中经硫脲关环制得去甲基氨噻肟酸乙酯(DMAT),再与α-溴代异丁酸叔丁酯在碱性条件下进行醚化反应,得到的醚化物在碱性条件下进行部分水解,得到头孢他啶侧链酸,然后与二硫二苯并噻唑(DM)在有机碱催化下反应得到头孢他啶侧链酸活性酯。

这条路线优点是原料乙酰乙酸乙酯价格便宜,醚化反应时间较短,采用价格 便宜的亚磷酸三乙酯代替价格昂贵的三苯基膦进行酯化反应。
5. 方法五
傅德才等可采用二氯甲烷及甲苯混合物作为反应溶剂,在碱性条件下,他啶酸与DM、三苯基膦反应,反应温度≤30 ℃,反应时间8 h,收率可达79.6%。 此法的缺点是甲苯与二氯甲烷的混合溶剂中,夹杂有少量未反应的头孢他啶侧链酸,不易于产物的纯化。
参考文献:
[1]杨晓辉. 头孢他啶侧链酸及其活性酯的合成研究[D].天津大学,2012.
[2]王庆全,唐伟.头孢他啶活性硫酯的合成研究[J].黑龙江医药,2009,22(04):495-496.
[3]王玉环.头孢他啶侧链酸活性硫酯的合成研究[J].石家庄职业技术学院学报,2009,21(02):33-35.
[4]梁宝臣,陈慧,杨俊杰.头孢他啶侧链酸及其活性硫酯的合成研究[J].中国抗生素杂志,2008(04):206-209.
[5]王玉环. 头孢他啶侧链酸及其活性硫酯的合成研究[D].北京化工大学,2004.


 1
1