本文重点介绍了梓醇红外光谱的预测方法,旨在为探讨梓醇及其衍生物的光谱、理化性质,以及环烯醚萜类化合物的化学结构和药理活性之间的关系提供有益的参考。
背景:地黄(Rehmanniag lutinosa Libosch)为玄参科多年生草本植物, 始载于《神农本草经》,是一种常见的中草药,广泛用于治疗多种疾病。 地黄中主要含有环烯醚萜苷类等物质,其中含量最高的是梓醇(Catalpol, Cat)。梓醇是发挥地黄药理作用的主要成分,也是地黄质量的控制指标。 大量研究表明,梓醇在改善心脑血管、增强免疫力、 抗炎症活性等方面, 有着良好的药理作用。
目前,对该类化合物的研究主要集中在化学结构、分布、药理作用以及提取分离方法等方面。然而,关于其分子结构、光谱特性以及理化性质等方面的理论研究相对较少。药物分子的药理活性与其分子结构密切相关,同一分子的不同构型或构象可能表现出不同的药理活性;同时,分子的振动光谱也是其重要性质之一,而量子化学则是研究这些性质的重要理论工具。密度泛函 (DFT) 考虑到电子交换和电子相关的能量, 使理论计算结果更精确, 研究表明DFT方法是探讨分子红外 (IR) 光谱的较好方法之一。
梓醇红外光谱的预测:
董春红等人采用DFT方法优化梓醇分子不同的构象异构体, 对最稳定的构象进行振动频率分析。具体如下:
(1)计算方法
梓醇分子的几何构型如图1所示,原子标号在图中标出。从分子结构上看,梓醇分子可以绕C(3)—C(13)和C(13)—C(15)单键旋转而形成不同的构象异构体。研究通过分别旋转单键C(3)—C(13)和C(13)—C(15)各120°得到不同的初始构象,采用DFT-B3LYP[15,16]方法在6-311G**水平分别对其进行几何优化,得到最稳定的构象;再用同样的方法和基组计算稳定构象的振动频率。用HF/6-311G**方法计算比较。
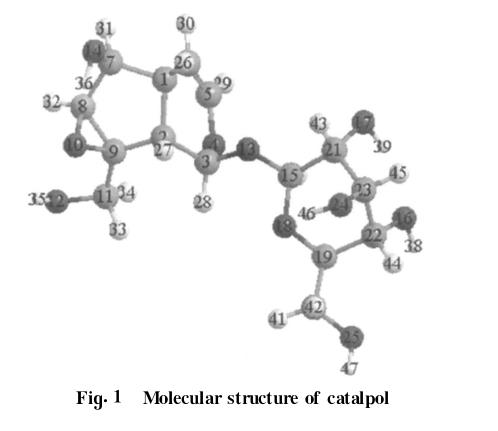
(2)梓醇的几何构型
分别对梓醇分子绕C(3)—C(13)和C(13)—C(15)单键旋转而形成的不同构象异构体进行理论计算。在计算过程中,通过改变二面角D1[C(3)—O(13)—C(15)—O(18)]和D2[C(15)—O(13)—C(3)—O(4)]得到九种不同的构象异构体。用B3LYP/6-311G**方法对九种不同的构象异构体进行几何构型全优化,得到最稳定的构象(即D1=-71.031°和D2=-66.056°)。
(3)梓醇的红外(IR)光谱
计算结果给出了梓醇分子的135种简正振动模式,没有虚频,说明得到的几何构型处于真正的位能面上极小点。频率的范围分布为3854~18cm-1,其中波数在500cm-1以上的振动模式有103个。用B3LYP/6-311G**方法计算出的梓醇分子的IR光谱如图2所示。
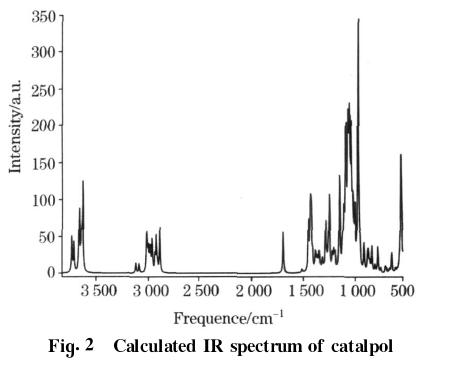
梓醇分子的振动频率在3700~500cm-1之间有5个IR特征吸收光谱区。3700~3500cm-1之间有6个强的吸收峰,分别属于6个O—H的伸缩振动。3200~2800cm-1之间有16个较强的吸收峰,分别属于16个C—H的伸缩振动。1715cm-1处的较强吸收属于CC的伸缩振动。1500~900cm-1之间属于C(O)—H的变形振动和C—O(C)的伸缩振动区,其中1500~1200cm-1之间的吸收峰属于C—H的变形振动,且多为混合振动形式,振动强度友弱有强;1200~900cm-1之间14个很强的吸收峰,分别属于C—O的伸缩振动,同时伴有C—H的变形振动。930cm-1处有个非常强的吸收,通过振动模式分析可以看到,O(4)—C(3)—O(13)—C(15)—O(18)—C(19)同时发生明显的振动,这与O—C在一般的结构中振动不显著的现象具有较大的差异,其可能的原因是这几种振动模式耦合的结果。900~500cm-1属于骨架振动的指纹区。
参考文献:
[1]秦兵,张伟东,李淑贤. 磁性表面分子印迹聚合物的制备及对梓醇的选择性吸附 [J]. 分析试验室, 2024, 43 (03): 394-401. DOI:10.13595/j.cnki.issn1000-0720.2023.012501.
[2]孙晓丽,董春红,孙雨安等. 基于密度泛函方法的梓醇红外光谱的预测 [J]. 光谱学与光谱分析, 2009, 29 (09): 2383-2387.


 1
1