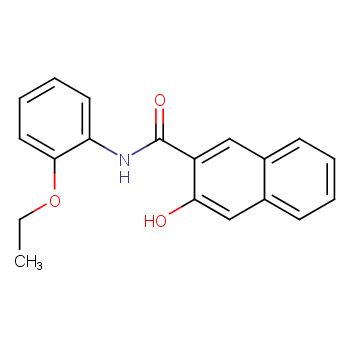
本文将讲述如何测定色酚AS-PH反应液中三种组分的含量,旨在为相关领域的研究人员提供参考依据。
背景:色酚AS-PH生产工艺多以邻氨基苯乙醚和2-羟基-3-萘甲酸为原料,在生产过程中,反应液中原料邻氨基苯乙醚和2-羟基-3-萘甲酸的转化率以及产品色酚AS-PH的收率是反应进程判断的 重要依据,对于工艺优化具有非常重要的意义。因此,色酚AS-PH工艺优化和实际生产过程中需要建立反应液中三种组分的测定方法。
目前,色酚AS-PH文献报道的检测方法主要用高效液相色谱法,其产品标准《HG/T 3959- 2007色酚AS-PH》中采用电位滴定法和高效液相色谱法测定色酚AS-PH的含量。检测2-羟基- 3-萘甲酸的方法主要有分光光度法和液相色 谱法,产品标准《HG/T 2745-2012 2-羟基-3-萘甲酸》中采用高效液相色谱-面积归一化法测定2- 羟基-3-萘甲酸及杂质含量。氨基苯乙醚类主要采用气相色谱法和气相色谱-质谱联用法测定。但这些方法都有一定的缺陷。
1. 合成:
(1)合成阶段
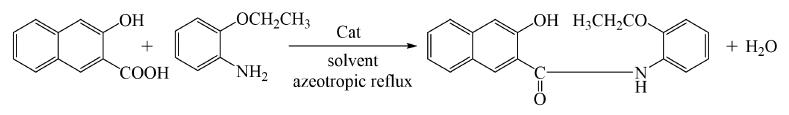
向装有温度计、冷凝管、分水器的250ml三口烧瓶中加入150ml氯苯,分水器中装有活化后的4?分子筛,开启搅拌,加入34.2g2-羟基-3-萘甲酸和一定量催化剂,加热至回流温度,2-羟基-3-萘甲酸和催化剂预反应1h,使用恒压漏斗缓慢滴加20.8g邻乙氧基苯胺到烧瓶中,保温反应60h。
(2)后处理阶段
反应结束进行降温,将反应液转移至单口瓶,使用旋转蒸发仪将溶剂氯苯脱除,然后用100ml无水甲醇溶解,进行抽滤浓缩,得一次粗产品,继续使用150ml 无水甲醇溶解滤饼,搅洗1h抽滤浓缩,用50ml无水甲醇淋洗,得精制湿产品。经真空105℃烘干3h,得到浅黄色粉末,产率94.4%,液相色谱纯度99%,熔点 157.2~157.9℃。
2. 检测:
邢伶等人建立了高效液相色谱法同时测定色酚AS-PH反应液中色酚AS-PH、邻氨基苯乙醚和2-羟基-3-萘甲酸含量的方法。色谱柱为Inert Sustain C18(4.6mm×150mm,5μm);检测波长为270nm;流动相为1g·L-1磷酸二氢 钾溶液(p H=3.0)-乙腈,梯度洗脱;流速为0.8mL·min-1,外标法测定三种组分含量。结果表明:色酚AS PH、邻氨基苯乙醚、2-羟基-3-萘甲酸与相邻色谱峰之间分离度良好,三种组分的浓度分别在2.0mg·L-1~ 498mg·L-1、2.0mg·L -1 ~494mg·L-1、1.0mg·L -1 ~249mg·L-1范围内线性关系良好,加标回收率分别为 97.3%~103.7%、96.6%~102.0%、101.2%~104.6%。该方法操作简便、准确度高,可以为色酚AS-PH的工艺优化和中控分析提供依据。实验方法为:
(1)色谱条件
色谱柱为Inert Sustain C18色谱柱(150mm× 4.6mm,5μm,日本岛津公司),柱温为35℃;进样量为5μL;检测波长为270nm;流动相A为乙腈,B 为1g·L-1磷酸二氢钾溶液,用磷酸调节pH值至 3.0;流速为0.8mL·min-1;梯度洗脱程序为:0~ 30min,10%A~90%A;30~35min,90%A;35.0~ 35.1min,90%A~10%A;35.1~40min,10%A。
样品测定采用色谱峰的保留时间定性,峰面积外标法定量。
(2)标准品溶液与样品溶液的配制
准确称取0.1g(精确至0.0001g)色酚AS-PH 标准品、0.1g邻氨基苯乙醚标准品和0.05g 2-羟基-3-萘甲酸标准品,加乙腈超声使溶解并定容至 100mL容量瓶中,作为混合标准储备液。
分别移取0.1、1.0、2.0、5.0、10.0、20.0、25.0mL混合标准储备液,用乙腈稀释定容至 50mL容量瓶中,配制色酚AS-PH、邻氨基苯乙醚浓度为2、20、40、100、200、400、500 mg·L-1,2-羟 基-3-萘甲酸浓度为1、10、20、50、100、200、250mg·L-1的混合标准溶液。所有标准储备液和混合标准溶液于20℃避光保存。
准确称取0.4g(精确至0.0001g)色酚AS-PH 反应液,置于100mL容量瓶中,用乙腈定容至刻度,摇匀,作为样品溶液。
所有溶液均用0.45μm尼龙滤膜过滤,滤液供检测用。
参考文献:
[1]邢伶,王瑞菲,唐晓婵等. 高效液相色谱法测定色酚AS-PH反应液中三种组分的含量 [J]. 化学研究与应用, 2020, 32 (12): 2281-2286.
[2]韩焕蓬. 色酚AS-PH合成新工艺的研究[D]. 青岛科技大学, 2020. DOI:10.27264/d.cnki.gqdhc.2020.000700


 1
1