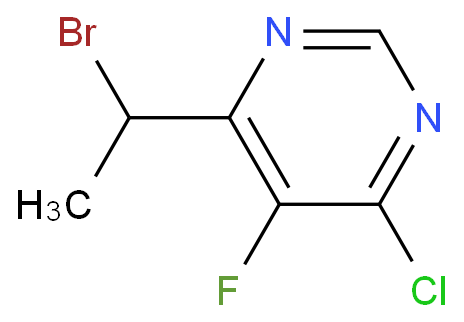
本文旨在介绍6-(1-溴乙基)-4-氯-5-氟嘧啶的合成方法,为进一步研究和开发新的应用提供了重要的参考依据。
背景:6-(1-溴乙基)-4-氯-5-氟嘧啶是合成新型抗真菌药物伏立康唑(voriconazole)的关键中间体。伏立康唑是由美国Pfizer公司2002年8月上 市的最新三氮唑类抗真菌药物。
合成:
1. 方法一:
以5-氟尿嘧啶为起始原料,经三氯氧磷氯化,溴化乙基镁格氏反应乙基化,经水解、还原、氯化,再经N-溴代琥珀酰亚胺溴化制得目标产物6-(1-溴乙基)-4-氯-5-氟嘧啶。总收率为51.6%。具体步骤如下:
(1)2,4-二氯-5-氟嘧啶(2)的合成
将92.0 g(0.6 mo1)三氯氧磷加入反应瓶中,搅拌下加入19.5 g(0.15 mol)5-氟尿嘧啶,升温到90℃,滴加36.5 g(0.5 mo1)N,N-二甲基苯胺,约1 h滴毕,溶液呈深褐色;升温至回流反应6 h,降至室温。反应液滴加到3 mol/L盐酸200 mL中,约1 h滴完,用3×50 mL二氯甲烷提取,合并有机层,再用4×50 mL水洗至中性,用无水硫酸钠干燥过夜,抽滤,有机层减压浓缩,得24.3 g红棕色油状物(2),收率:97.1%。
(2)2,4-二氯-6-乙基-5-氟嘧啶(3)的合成
将12.1 g(0.5 mol)镁屑及120 mL四氢呋喃(THF)加入反应瓶中,于40℃以下搅拌,滴加 54.5 g(0.5 mol)溴乙烷的50 mL THF溶液,滴毕,反应1 h,降温至0℃,滴加41.7 g(0.25 mol)2的 THF溶液100 mL,滴加过程中保持温度15℃以下,滴毕,冷却至0℃,依次滴加34 g(0.33 mol)三乙胺,及85 g(0.67 mol)碘的THF溶液200 mL,滴加过程中保持温度5℃以下,0℃反应2 h,滴毕,滴加水200 mL,保持温度10℃以下,用3 mol/L 盐酸调pH至1,用甲苯2×250 mL提取,然后4× 500 mL水洗有机相,无水硫酸钠干燥过夜,抽滤, 减压蒸干得42.1 g浅黄色油状物(3),收率86.3%。
(3)2,4-二氯-6-乙基-5-氟嘧啶-4-酮(4)的合成
将39 g(0.2 mol)3、2 mol/L氢氧化钠溶液 100 mL加入反应瓶中,加热至80℃反应2.5 h,降至室温,将反应液倒入200 mL二氯甲烷中,滴加浓盐酸调pH至3,分出有机层,减压蒸干,加入乙酸乙酯150 mL,回流30 min,过滤,滤液析出 晶体,过滤,抽干,得32.3 g淡黄色晶体(4),收率91.7%,m.p.101~104℃。
(4)6-乙基-5-氟-4-羟基嘧啶(5)的合成
将26.5 g(0.15 mol)产物4,100 mL乙醇及 5 mL水,再加入25 g(0.3 mol)醋酸钠及2.72 g(0.025 mol)5%钯碳加入反应瓶中,搅拌下在50℃ 和3×105Pa条件下氢化5 h,加入10 mL水终止反应,过滤,滤液减压蒸除乙醇,加入2×50 mL二 氯甲烷提取,合并有机层,减压蒸干,加入50 mL 甲苯,0℃搅拌3 h,过滤,得到16.8 g类白色晶体 (5),收率79%,m.p.110~113℃。
(5)6-乙基-4-氯-5-氟嘧啶(6)的合成
将21.3 g(0.15 mol)产物5,60 mL二氯甲烷,15.2 g(0.15 mol)三乙胺及25.3 g(0.23 mol) 三氯氧磷加入反应瓶中,搅拌下加热回流5 h,降温至20℃以下,倒入80 mL3mol/L盐酸中,分出有机层,水层用2×50 mL二氯甲烷提取,合并有机层,4×50 mL水洗,无水硫酸钠干燥过夜,抽滤,减压蒸干,得22.8 g浅红色油状物6,收率 95.2%。
(6)6-(1-溴乙基)-4-氯-5-氟嘧啶(1)的合成
将32.1 g(0.2 mol)化合物6,1.6 g(0.01 mol)偶氮二异丁腈(AIBN),53.4 g(0.3 mol)NBS及150 mL氯仿加入反应瓶中,光照,通氮气片刻,搅拌下加热至回流,反应6 h,冷却至 25℃,加水150 mL,分出有机层,水层用2×60 mL 氯仿提取,合并有机层,2×100 mL5%焦亚硫酸钠溶液洗,无水硫酸钠干燥过夜,抽滤,减压抽干,得42.8 g淡黄色油状物1,收率89.5%。
2. 方法二:
具体实验步骤如下:
(1)α-氟丙酰乙酸乙酯(2)的合成
1 000 mL三颈瓶中通入氮气保护,加入无水乙醚400 mL、乙醇钠22.44 g(0.34 mol),室温下磁力搅拌,缓慢滴加氟乙酸乙酯35.34 g(0.34 mol)。滴完后室温反应3 h,冰浴,取丙酰氯31.2 g(0.34 mol),混于50 mL无水乙醚中,缓慢滴加,回流4 h后倒入冰水中,取醚层,无水硫酸钠干燥,减压蒸馏,收集105~107℃/4.27 kPa馏分,得无色液体35.1 g,收率65%。
(2)6-乙基-5-氟-4-羟基嘧啶(3)的合成
冰浴下,于250 mL单颈瓶中加入12.96 g(0.08 mol)化合物2、甲醇钠-甲醇溶液50 mL(8.64 g,0.16 mol)、甲脒乙酸盐8.32 g(0.08 mol),0℃下反应1 h。室温反应过夜,再回流30 min,自然冷却,用冰醋酸调pH 6~7,加入乙酸乙酯-丙酮(V∶V=1∶3),提取若干次,取乙酸乙酯层浓缩,冰箱放置得到白色晶体,干燥后得8.9 g,收率78%,mp 107~110℃。
(3)4-氟-6-乙基-5-氟嘧啶(4)的合成
取2 g(14 mmol)化合物3、二氯甲烷10 mL、三乙胺2 mL溶于50 mL单颈瓶中,冰浴使内温低于40℃,滴加POCl3,回流5 h。冷却至室温,冰浴,缓慢滴加3 mol/L盐酸8.8 mL,取有机层,干燥蒸去二氯甲烷,得浅黄色油状物2 g,收率88.5%。
(4)6-(1-溴乙基)-4-氯-5-氟嘧啶(1)的合成
将1.9 g(12 mmol)化合物4、偶氮异丁腈(AIBN)0.1 g(0.1 mmol)、NBS 2.45 g(13.7 mmol)和二氯甲烷10 mL溶于反应瓶中,通入氮气保护,回流12 h,冷至室温,加水12 mL,水层用二氯甲烷提取,合并有机层,用0.96 mol/L亚硫酸氢钠溶液洗有机层,浓缩得油状物2.6 g,收率93%。
参考文献:
[1]李雪妍,刘媛媛,凌婷婷. 6-(1-溴乙基)-4-氯-5-氟嘧啶的合成工艺研究 [J]. 精细化工中间体, 2013, 43 (01): 25-26+29. DOI:10.19342/j.cnki.issn.1009-9212.2013.01.007.
[2]姚斌,程斌,吴秋业. 6-(1-溴乙基)-4-氯-5-氟嘧啶合成新方法 [J]. 中国药物化学杂志, 2004, (01): 51-52.


 1
1