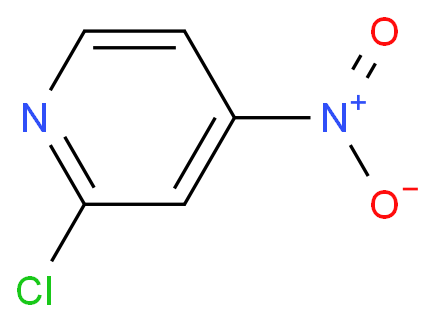
本文将探讨如何合成2-氯-4-硝基吡啶的方法,使读者将能够更全面地了解该化合物的制备过程。
背景:吡啶基苯脲类植物生长调节剂、除草剂和吡啶基医药产品在我国已得到广泛的认可和应用,2-氯-4-硝基吡啶(CNP)是合成它们的重要中间体。
合成:
1. 方法一:
(1)吡啶-N-氧化物的合成
在装有电搅拌器、温度计和滴液漏斗的500 mL三口瓶中,加入吡啶40 g(0.5 mol)、乙酸200 mL和45%过氧化氢60 mL,混合物在70~80 ℃水浴上加热3 h,再滴加过氧化氢30 mL,在相同温度下加热9 h,减压浓缩混合物至100 mL左右,加水100 mL稀释,再减压蒸馏除去水,残留物用无水碳酸钠中和至强碱性,用200 mL氯仿振荡萃取,静置滤除碳酸钠和乙酸钠沉淀。滤液用无水硫酸钠干燥,蒸除氯仿,高真空减压蒸馏,,收集100~103 ℃/133 Pa 的馏份,得无色固体46 g,产率为96%,熔点为65~ 66 ℃。
(2)4-硝基- N-氧化吡啶的合成
将10 g氧化吡啶溶在12 g浓硝酸(1.48)和30 mL浓硫酸的混合液中?在油浴中于128~130 ℃加 热3~5 h?冷却后倒到碎冰中?分小量加固体碳酸 钠?当晶体沉淀后?过滤?并用冰水洗涤?滤液碱化后 用氯仿抽提?可再得一些产物?两次粗产物合并?以水重结晶,得 10.6 g,熔点为 159 ℃。
(3)2-氯-4-硝基吡啶的合成
将5g 4-硝基-N-氧化吡啶加入到 100 mL的三口瓶中,在搅拌下加入 75mL的POC13,慢慢升温到 50~60℃,反应3h后再升温到115~120℃,反应 1.5 h 后,蒸除,回收POC13。将固体剩余物倒入到 400 mL 的冰水中,并快速搅拌,冷却后用NaOH 调溶液的 pH 值,放入冰箱过夜,得到黄色沉淀后,过滤、水洗、干燥,得产品 3.2 g,熔点为53℃。
2. 方法二:
(1)2-氯吡啶-N-氧化物的合成
在500 ml 三口反应器中分别加入冰醋酸、2-氯吡啶、浓硫酸和30%过氧化氢,在反应温度下保温搅拌3 h,冷却,用20%氢氧化钠溶液中和至中性,减压蒸馏回收未反应的2-氯吡啶,残液毋需处理直接进行下步反应。
(2)2-氯-4-硝基吡啶-N-氧化物的合成
用冰浴将上述残液冷至2℃ ~3℃ ,搅拌下加入事先冷至5℃以下的计量浓硫酸,于2℃ ~3℃滴加浓硫酸和90%发烟硝酸的混合物(60 min内滴加完 毕),之后,慢慢升温至90℃,保温搅拌4 h~6 h。
将反应混合物冷至10℃,倾入适量的冰-水混合物中,用碳酸钠中和、过滤,棕褐色滤饼即为2-氯-4-硝基吡啶-N-氧化物产品,空气中晾干后,溶于500 ml热氯仿中。滤液用100 ml氯仿萃取2次,萃取液合并于上述氯仿溶液中,蒸去氯仿,得棕褐色固体产物2-氯-4-硝基吡啶-N-氧化物。
(3)2-氯-4-硝基吡啶的合成
上述所制2 -氯-4-硝基吡啶-N-氧化物,用乙酸乙酯溶解,滴加一定量的三氯化磷,于反应温度下加热搅拌20 min~30 min。冷至室温,倾入冰-水混合物中, 用20%氢氧化钠溶液中和至中性,用氯仿萃取,萃取液用硫酸镁干燥,过滤,蒸去溶剂,用氯仿-乙醇重结晶,得浅黄色针状固体即为纯品2-氯-4-硝基吡啶产物。
参考文献:
[1]李建康,周彦水,陈龙. 2-氯-4-硝基吡啶的制备 [J]. 河北化工, 2001, (03): 20-23.
[2]王长守,黄建良,张志杰. 2-氯-4-硝基吡啶的合成 [J]. 陕西化工, 1999, (01): 17-18+34. DOI:10.16581/j.cnki.issn1671-3206.1999.01.007


 1
1