本文旨在介绍合成硫酸头孢噻利的方法,通过深入探讨硫酸头孢噻利的合成过程,我们旨在为其制备提供可靠的实验方法和理论基础。
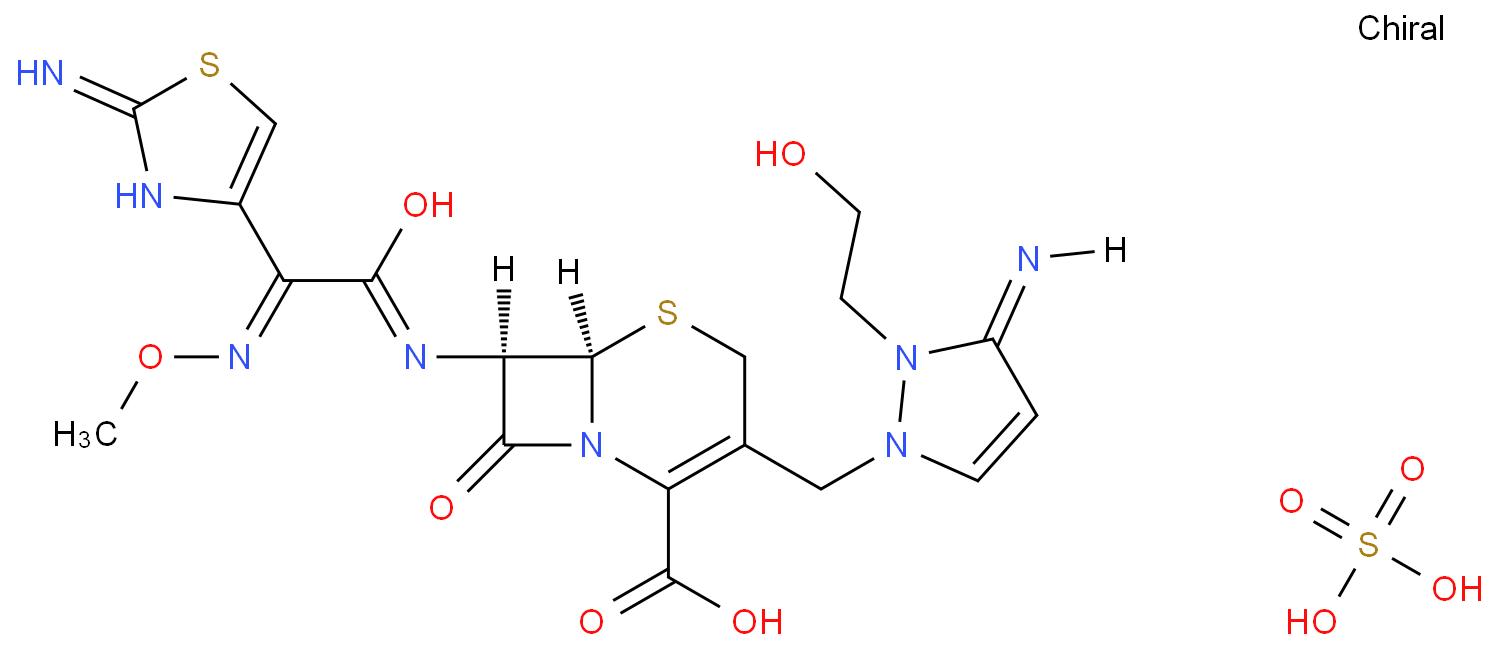
简述:硫酸头孢噻利 (cefoselis,FK-037) 是日本藤泽药品工业株式会社和美国Johnson公司共同研制开发的第四代注射头孢菌素,其化学名为7β-[ (z) -2- (2-氨基-4-噻唑基) -2- (2-甲氧亚氨基乙酰氨基) ]-3-[3-氨基-2- (2-羟乙基) -1-吡唑鎓基]甲基-3-头孢烯-4-羧酸硫酸盐,1998年首次在日本上市。硫酸头孢噻利对临床常见的大多数革兰阴性菌和阳性菌具有良好的抗菌活性。其抗菌活性的主要优势是对甲氧西林耐药性金黄色葡萄球菌及假单胞菌有良好的抗菌活性,尤其是对铜绿假单胞菌的活性,而目前其他头孢菌素类抗生素对这些细菌活性较差。
合成:
1. 方法一
以3-氯甲基-7-叔丁氧酰基氨基-3-头孢烯-4-二苯甲基羧酸酯为起始原料,与5-甲酰氨基-1-(2-甲酰氧乙基)吡唑化合,然后经过三氟乙酸、浓盐酸等步骤去保护基,得到7-氨基-3-[3-氨基-2-(2-羟乙基)-1-吡唑甲基]-3-头孢烯-4-羧酸盐酸盐,经提纯后与氨噻肟酸苯并三唑酯反应得到硫酸头孢噻利。该方法采用水作溶剂,改进了提纯工艺,总收率为18.8%。

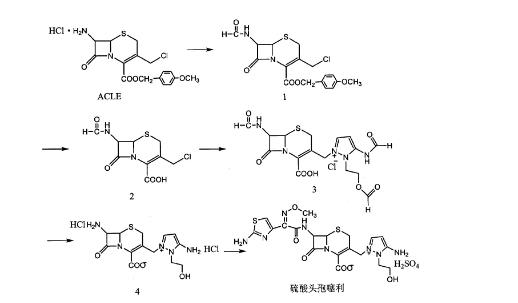
2. 方法二
以7β-烷酰氨基-[3-烷酰氨基-2- (2-烷酰氧乙基) -1-吡唑鎓基]甲基-3-头孢烯-4-羧酸盐化合物 (ACLE) 为原料,经上保护,酯解,上三位,水解后,与α- (2-氨基噻唑-4-基) - α- (Z) -甲氧亚氨基乙酸 (苯并噻唑-2-基) 巯基酯 (简称AE活性酯) 反应,制得硫酸头孢噻利。
3. 方法三
以7-氨基头孢烷酸为原料采用“一锅煮”的方法合成关键中间7β-胺基-3-[3-胺基-2- (2-羟乙基) -1-吡唑嗡甲基]-3-头孢烯-4-羧酸盐酸盐,再与2- (2-氨基-4-噻唑基) -2- (甲氧亚氨基) 乙酸硫代苯并噻唑酯缩合得到头孢噻利。具体步骤如下:
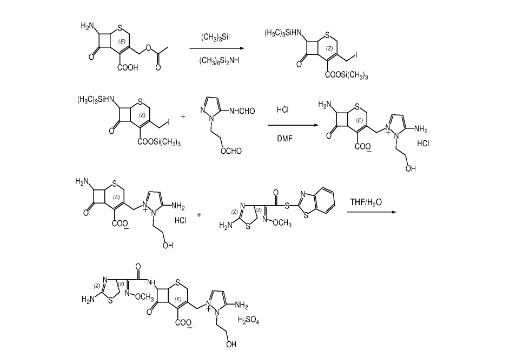
(1)7β-胺基-3-[3-胺基-2- (2-羟乙基) -1-吡唑嗡甲基]-3-头孢烯-4-羧酸盐酸盐的合成
在配有搅拌器、温度计、氮气导管及恒压滴液漏斗的2000mL四口烧瓶中,加入80g (0.295mo1) 7-氨基头孢烷酸 (7-ACA) 、800g二氯甲烷,搅拌冷却至10℃左右。搅拌下加入55g (0.34mo1) 六甲基二硅烷胺、1g三甲基碘硅烷、1g N,N-二甲基甲酰胺,然后自然升到室温,在室温下搅拌反应5~12h。然后降温到-5℃左右,在N2保护下,滴加三甲基碘硅烷125g (0.625mol) ,滴加完毕,反应温度控制在0~-5℃,搅拌反应2h。在此温度范围内,慢慢分批加入,80g (0.44mol) 5-甲酰胺基胺基-1- (2-甲酰氧乙基) 吡唑。在0℃下搅拌反应过夜。第二日缓慢升温至l4℃,搅拌反应2.5h,得到棕青色透明溶液。在5000mL三口烧瓶中,加入60g的DMF、300g浓盐酸、300g水,搅拌冷却至0℃,将上述棕青色透明溶液缓慢加入该混合液中,搅拌反应5h。然后分液,弃去有机相收集水相,搅拌下,向水相,缓慢加入1500g丙酮,产生大量的晶体。搅拌约1h后,冰水冷却,抽滤,滤饼用丙酮淋洗,真空干燥,得到白色晶体,即7β-胺基-3-[3-胺基-2- (2-羟乙基) -1-吡唑嗡甲基]-3-头孢烯-4-羧酸盐酸盐83.5g,收率75.9%。
(2)硫酸头孢噻利的合成
在500mL三口烧瓶中,加入80g四氢呋喃和80g水,搅拌冷却至0℃。加入80g (0.216 mol) 7β-胺基-3-[3-胺基-2- (2-羟乙基) -1-吡唑嗡甲基]-3-头孢烯-4-羧酸盐酸盐、80g (0.227 mo1) AE活性酯,在搅拌下,于20~25min内滴加22 g (0.216 mol) 三乙胺调节体系p H到9.0,滴加过程温度不能超过5℃,加完之后,搅拌30min。缓慢升温至25℃左右,再搅拌反应4h。反应液用200g乙酸乙酯分两次萃取,合并水相,用活性炭脱色。在5℃左右,溶液用20%硫酸调节p H值至1~2。在搅拌下,向滤液中滴加异丙醇至出现大量的晶体,继续搅拌1h。冷却、抽滤,用异丙醇淋洗,常温减压干燥,得到白色晶体即硫酸头孢噻利117g,收率87.5%。
参考文献:
[1]崔德修,李重阳,李明研. 硫酸头孢噻利的合成 [J]. 国外医药(抗生素分册), 2013, 34 (06): 250-251+256. DOI:10.13461/j.cnki.wna.005022.
[2]王亚江,李玉荃,刘志友等. 硫酸头孢噻利的合成 [J]. 天津药学, 2010, 22 (04): 75-76.
[3]薛峰,居沈贵,姚虎卿. 硫酸头孢噻利合成工艺研究 [J]. 中国新药杂志, 2005, (03): 322-324.


 1
1