本文介绍了如何使用(E)-3-[3-(4-氟苯基)-1-异丙基吲哚-2-基]丙烯醛来合成氟伐他汀钠。
背景:(E)-3-[3-(4-氟苯基)-1-异丙基吲哚-2-基]丙烯醛是氟伐他汀钠的重要合成中间体。氟伐他汀钠是一种通过化学合成得到的降血脂新药,作为3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂,是继洛伐他汀、普伐他汀和辛伐他汀之后开发的第4个同类药物。氟伐他汀最早由瑞士Sandoz公司开发,在1993年投产并于1994年1月首次在英国上市,随后逐渐在美国、加拿大、新西兰等多个国家上市,并于1997年在我国注册。
氟伐他汀是一种强效降脂药物,无需代谢转化即具有药理活性。其分子中的氟苯吲哚部分模拟辅酶的结构,与HMG-CoA还原酶相互作用,竞争性地抑制该酶催化HMG-CoA转化为甲羟戊酸的关键步骤,从而减少细胞内胆固醇积累,增加低密度脂蛋白胆固醇(LDL-C)受体数量,恢复胆固醇代谢平衡,并促进LDL-C的清除。氟伐他汀为右旋和左旋两种消旋体组成,其中右旋活性强30倍。使用氟伐他汀能够降低血浆总胆固醇、甘油三酯和低密度脂蛋白,同时增加高密度脂蛋白水平,并抑制动脉血管粥样硬化的发展过程,通过抑制甲羟戊酸还原酶来抑制动脉肌细胞的增生和生长。
(E)-3-[3-(4-氟苯基)-1-异丙基吲哚-2-基]丙烯醛合成氟伐他汀钠的路线:
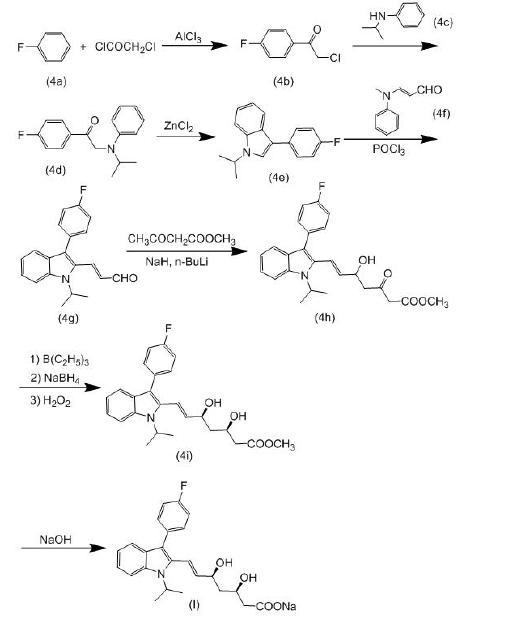
(1) 根据1988年美国专利4739073的公开内容,提供了一种合成(3R,5S)-氟伐他汀钠的方法。该方法以氟苯(4a)为起始原料,与氯乙酰氯反应形成4-氯乙酰氟苯(4b)。然后将化合物(4b)与氮异丙基苯胺(4c)进行缩合反应生成中间体(4d),再经过无水氯化锌催化下的环合反应形成吲哚环,得到化合物(4e)。接下来,将化合物(4e)与3-N-甲基-N-苯基胺基丙烯醛(4f)进行Vilsmeier-Haack反应,得到(E)-3-[3'-(4"-氟苯基)-1'-(1"-甲基乙基)吲哚-2'-基]-2-丙烯醛(4g)。然后,化合物(4g)与乙酰乙酸甲酯缩合生成β-羟基酮,随后使用三乙基硼烷和硼氢化钠作为还原剂将羰基还原为羟基,最后通过氢氧化钠的水解反应得到目标产物(I)(3R,5S)-氟伐他汀钠。
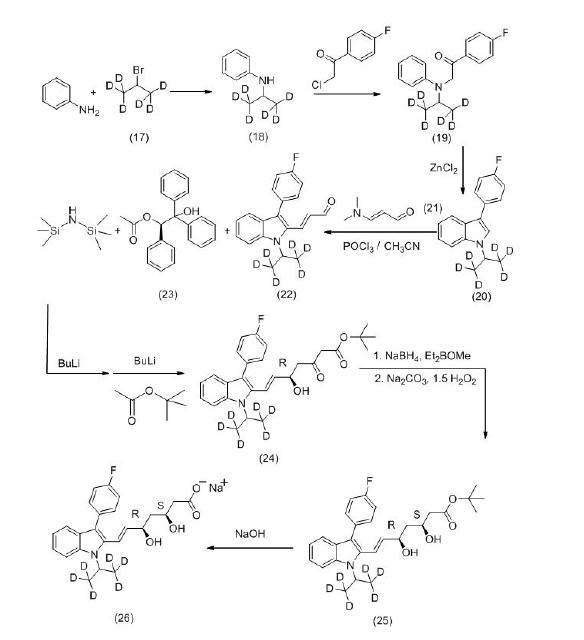
(2)以苯胺和含有6个氘原子的2-溴丙烷(17)为原料,生成N-异丙基苯胺-d6(18),再与4-氟-苯乙酰氯缩合生成化合物(19),经分子内环合生成化合物(20),再与N,N-二甲基 丙烯醛(21)发生Vilsmeier-Haack反应,生成( E ) -3-[ 3′-( 4″-氟苯基) -1′- (异丙基[d6])-吲哚-2′-基]-2-丙烯醛(22)。使用(R)-2-羟基-1,2,2-三苯基乙酸乙酯(23)作为手性催化剂,将中间体(22) 直接转化为5位具有所需要合成的R构型羟基的化合物(24)。使用syn-选择性还原剂 Et2BOMe/Na BH 4将化合物(24)中3位羰基选择性还原为羟基,直接得到所需要(3S,5R)构型的氟伐他汀叔丁酯-d6,经氢氧化钠水解得最终产物(3S,5R)-氟伐他汀钠-d 6,反应路线如图。
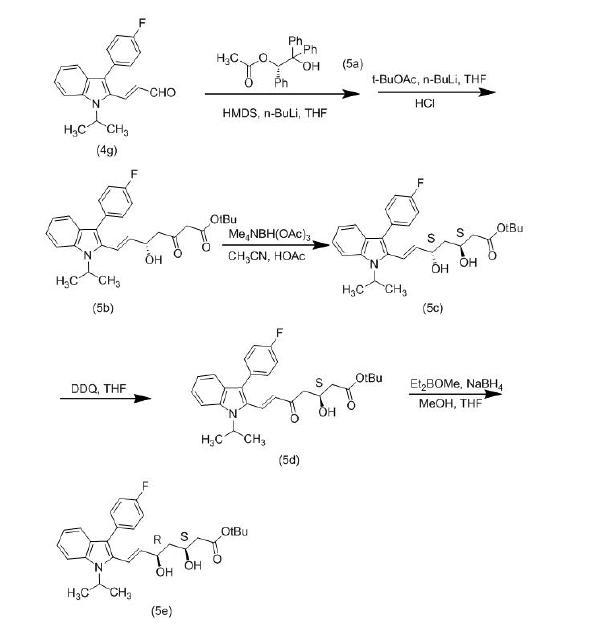
(3)根据Orin Tempkin等人于1997年在Tetrahedron发表的论文,研究人员采用了一种新的合成路线来制备(3S,5R)-氟伐他汀钠。首先,在合成中间体(4g)之后,使用手性催化剂(S)-2-羟基-1,2,2-三苯基乙酸乙酯(5a),将不饱和醛转化为具有相同构型的β-羟基酮酯,并与乙酸叔丁酯反应生成化合物(5b)。这个反应可以在同一个容器中进行,并具有高度的选择性。通过简单的重结晶处理,可获得约98%的对映体纯度。接下来,关键的中间体β-二酮可以经由以下步骤合成(3S,5R)-氟伐他汀钠。首先,研究人员使用抗选择性还原剂Me4NHB(OAc)3对化合物(5b)进行还原得到(3S,5S)-氟伐他汀叔丁酯(5c)。然后,他们使用DDQ将5位羟基氧化为酮(5d)。最后,通过使用syn-选择性还原剂Et2BOMe/NaBH4将5位还原为R构型的羟基,得到所需的(3S,5R)-氟伐他汀叔丁酯(5e)。通过简单的水解处理,即可得到目标化合物(3S,5R)-氟伐他汀钠。
参考文献:
[1]吴科颖. 氘标记药物标准品双氯芬酸、氟伐他汀钠的合成[D].南京航空航天大学,2010.


 1
1

![(E)-3-[3-(4-氟苯基)-1-异丙基吲哚-2-基]丙烯醛化学结构式](https://structimg.guidechem.com/9/9/28568.png)