提问
问答首页
问题分类
文章
视频
消息管理
站内通知
我的
盖德化工网
盖德化工网
>
盖德问答
>
新药杂质质量控制?
化药
工艺技术
新药杂质质量控制?
本帖内容被屏蔽
0评论
+
关注
共4个回答
勼欢孤欢,电气(高压)
2019-01-20回答
创新药的杂质标准本来就是临床完成才最终确定标准的,至于临床前标准订多少就要评估风险了,太大可能会对安评临床不利,太小可能又对工艺制造难度,不容易达到。超过0.15%可以申报临床的,不过最好还是要结构鉴定,临床前研究很大一块就是安全性评价,安评结果通过了就没问题了,最后临床完成后可以将杂质标准缩紧以限制以后的仿制药。
10
评论 0
举报
明天.方宇.,质量保障部副经理
2019-01-20回答
分析杂质来源,0.15%应该有结构确证了,分析是否是基因毒性,因为是新药杂质限度还是取决于临床结果,治疗作用于0.15%杂质有无关系都是个问号。
3
评论 0
举报
也好,自控设计工程师
2019-01-20回答
建议看一下杂质研究的指导原则,说的很清楚的。
没有一个说法是杂质超过了多少就不让报临床。只是从安全行的角度出发,或者要求你去证实他的安全性。并且在这个基础上,尽量将杂质控制的低一点。
展开
11
评论 0
举报
宝~~贝,化工研发
2019-01-20回答
首先你要确定你的0.15%杂质是工艺杂质还是降解杂质?
工艺杂质的话,鉴别后,分析其来源,争取在合成工艺中减少或者除去;
降解杂质的话,通过破坏试验,确定主要降解途径是什么?在稳定性实验中是否增加?增加到多少?鉴别后,查阅文献或者通过实验说明超过限度的理由。
展开
15
评论 0
举报
上一条:
曲格列汀琥珀酸盐质量标准求助?
下一条:
求助USP38、BP2015 Clonidine Hydrochloride原料及制剂相关标准?
杂质
CAS No:1886-34-6
国内供应商(25家)
深圳振强生物技术有限公司
VIP
10年
备案
试剂
联系电话:
0755-66853366
产品名称:
盐酸硫胺素杂质12
价格:
电询
主营产品:
莫匹罗星EP/USP杂质F;莫匹罗星杂质F、苯肾上腺素异构体杂质;苯肾上腺素S异构体,S-苯肾上腺素、托法替尼(3S,4R)异构体;非对应异构体CP-733,315、多西他赛杂质14(多西他赛巴豆醛类似物)、
在线询盘
洽谈
湖北扬信医药科技有限公司
VIP
10年
备案
联系电话:
产品名称:
硫胺素杂质维生素B1杂质)1886-34-6 现货
价格:
电询
主营产品:
伐地那非杂质31 1339092-18-0、阿齐沙坦杂质(Azilsartan)全套汇总分享、树脂毒素(RESINIFERATOXIN)57444-62-9、O-丙基伐地那非 2840532-32-1、
在线询盘
洽谈
上海源叶生物科技有限公司
VIP
12年
备案
品牌试剂
联系电话:
18021061708-聂先生
产品名称:
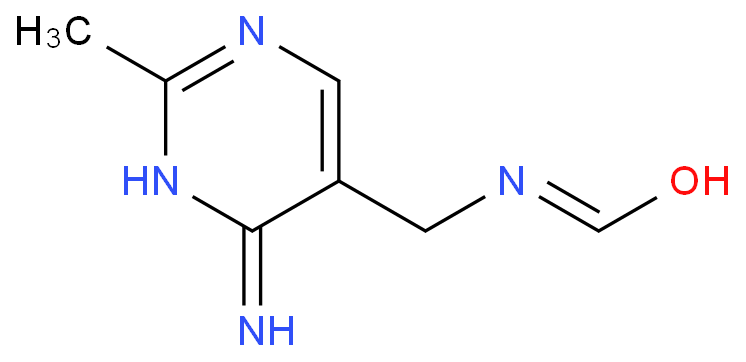
4-Amino-5-(formamidomethyl)-2-methylpyrimidine
价格:
3600元 /瓶
主营产品:
DEAE纤维素DE-52、尿苷二磷酸葡糖醛酸、羧苄青霉素钠、胆碱氧化酶、
在线询盘
洽谈
上海洽姆分析技术有限公司
VIP
10年
备案
联系电话:
86-021-52969808
产品名称:
4 -氨基-5-(formamidomethyl)- 2 - methylpyrimidine
价格:
99元 /瓶
主营产品:
Tenax GR 吸附剂、CG1TD6080 Carbograph 色谱填料、20273 Carbopack石墨化碳黑吸附剂 和 Carboxen 碳分子筛吸附剂(气相色谱填料)、11982 Tenax TA 多孔聚合物吸附剂 60-80目、
在线询盘
洽谈
湖北扬信医药科技有限公司
VIP
9年
备案
联系电话:
产品名称:
硫胺素杂质维生素B1杂质)1886-34-6 现货
价格:
电询
主营产品:
软毛青霉酸99-23-0现货供应、达泊西汀全套杂质现货供应119356-77-3、洛索洛芬开环杂质1091621-61-2 现货供应、他克莫司一水合物(Tacrolimus monohydrate)109581-93-3 现货供应、
在线询盘
洽谈
查看更多供应商 >
杂质相关回答
关于非特定降解杂质、特定降解杂质、过程杂质如何定义和理解?
4个回答
关于降解杂质?
1个回答
药物杂质研究?
6个回答
杂质含量标定?
7个回答
杂质验证问题?
4个回答
您可能感兴趣的问答
三价铬溶液颜色问题?
15个回答
能否用离心代替旋蒸去除乙醇?
2个回答
想请教下靛蓝染料在紫外分光光度计下吸收的问题?
4个回答
硫酸钙结垢,用什么清洗掉?
4个回答
氰基取代苯环上的卤素的反应条件?
0个回答
请填写举报原因
选择举报原因
垃圾广告
有害信息
文不对题
涉嫌侵权信息
其他
增加悬赏
剩余能量值
能量值
提示
提示信息
确定
取消
消息提示